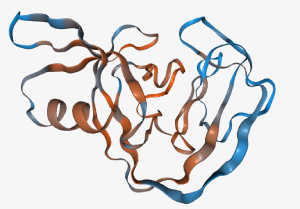
 The structure is colored according to the probability that the split sites are active (blue) and inactive (red).
The structure is colored according to the probability that the split sites are active (blue) and inactive (red).
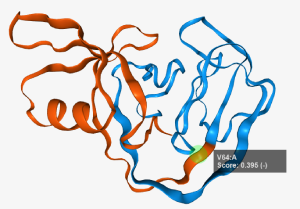 Residues can be selected via Ctrl+Leftclick (a green sphere will show the selection) which treat the selected residue as the site where the protein is split. The split occurs after the selected residue.
Additionally the two split fragments are shown with two colors (red and blue).
Selected residues can be deselected via Ctrl-Leftclicking anywhere but on a residue. A tooltip will appear when
hovering the mouse over the residue. The residues' chain information is displayed after a colon.
Residues can be selected via Ctrl+Leftclick (a green sphere will show the selection) which treat the selected residue as the site where the protein is split. The split occurs after the selected residue.
Additionally the two split fragments are shown with two colors (red and blue).
Selected residues can be deselected via Ctrl-Leftclicking anywhere but on a residue. A tooltip will appear when
hovering the mouse over the residue. The residues' chain information is displayed after a colon.
 The current camera view can be saved via this button.
The current camera view can be saved via this button. The size of the structure window can be resized via this button.
The size of the structure window can be resized via this button.
 Different groups of split sites can be selected outputting a list of split sites ordered from the highest activity probability to lowest.
The high active probability group encompasses sites with a activity probability of at least 0.75. The high inactive probability encompasses sites with a activity probability of less than 0.25.
The active and inactive groups have all sites that the model would predict to be active (at least 0.5) and inactive (less than 0.5) respectively.
Different groups of split sites can be selected outputting a list of split sites ordered from the highest activity probability to lowest.
The high active probability group encompasses sites with a activity probability of at least 0.75. The high inactive probability encompasses sites with a activity probability of less than 0.25.
The active and inactive groups have all sites that the model would predict to be active (at least 0.5) and inactive (less than 0.5) respectively.
 The list can be copied to the clipboard with this button.
The list can be copied to the clipboard with this button.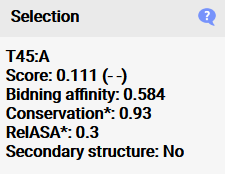 When a residue is selected, additional information concerning the conservation,
binding affinity, surface accessibility and secondary structure properties are shown here which are used to calculate the activity probability in the model. It is important to note that
conservation and surface accessibility are given as means of the surrounding residues of the split site, as this was shown to work best for predicting the activity,
so the values do not show single residue values (which can be accessed in the raw files) (*). The split site is considered to be in a secondary structure if both flanking residues of the split site are in
a secondary structure region.
When a residue is selected, additional information concerning the conservation,
binding affinity, surface accessibility and secondary structure properties are shown here which are used to calculate the activity probability in the model. It is important to note that
conservation and surface accessibility are given as means of the surrounding residues of the split site, as this was shown to work best for predicting the activity,
so the values do not show single residue values (which can be accessed in the raw files) (*). The split site is considered to be in a secondary structure if both flanking residues of the split site are in
a secondary structure region.
 Deselecting residues can be done in the structure view or with this button located at the top of the Data tab.
Deselecting residues can be done in the structure view or with this button located at the top of the Data tab. If multiple files were uploaded via batch upload, the single files can be selected via a dropdown menu.
If multiple files were uploaded via batch upload, the single files can be selected via a dropdown menu. Zooming out can be done via a double-click on the graph or via this button that appears at the top-left of the graph (if it is zoomed in).
Zooming out can be done via a double-click on the graph or via this button that appears at the top-left of the graph (if it is zoomed in).
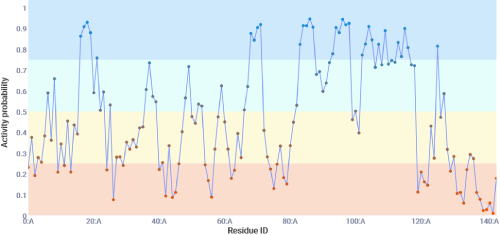 This tab shows the models prediction for each possible split site. There are 4 categories given with 4 colors.
Generally speaking the model predicts split sites with a probability of at least 0.5 to be active and below 0.5 to be inactive. Sites with a probability of at least 0.6 have been shown to
contain the highest ratio of true positives to false positives at the dispense of loosing some true positive split sites. Sites with a probability of less than 0.4 have been shown to
contain the highest ratio of true negatives to false negatives.
This tab shows the models prediction for each possible split site. There are 4 categories given with 4 colors.
Generally speaking the model predicts split sites with a probability of at least 0.5 to be active and below 0.5 to be inactive. Sites with a probability of at least 0.6 have been shown to
contain the highest ratio of true positives to false positives at the dispense of loosing some true positive split sites. Sites with a probability of less than 0.4 have been shown to
contain the highest ratio of true negatives to false negatives.
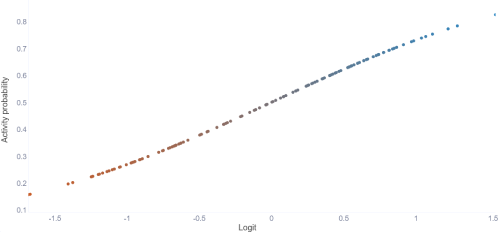 This tab shows the logit function, which what the model's prediction is based on. The points are sorted by the lowest prediction to the highest (Meaning the splits are not in order like with the other tabs).
This tab shows the logit function, which what the model's prediction is based on. The points are sorted by the lowest prediction to the highest (Meaning the splits are not in order like with the other tabs).
 The sequence tab shows the sequence with the activity probability color codes,
residue id's and whether the residue belongs to a secondary structure region (marked with Sec).
The sequence tab shows the sequence with the activity probability color codes,
residue id's and whether the residue belongs to a secondary structure region (marked with Sec).
 The raw data can be downloaded in this tab as a zip file.
The zip contains the output from DSSP, hmmer, muscle and rate4site as well as the pdb file that was generated through pdb2pqr at a pH of 7.
The raw data can be downloaded in this tab as a zip file.
The zip contains the output from DSSP, hmmer, muscle and rate4site as well as the pdb file that was generated through pdb2pqr at a pH of 7.
Int&in, as the name easily reveals, is dedicated to inteins. In particular, split inteins.
Inteins are "intervening proteins": proteins found within other proteins, albeit transiently. As a matter of fact, inteins eventually excise themselves from their precursors in a process known as protein splicing (Hirata et al., 1990; Kane et al., 1990). This is an autocatalytic process, meaning that it does not need any co-factor, which concludes itself with the formation of a peptide bond between the two polypeptides initially bordering the intein (referred to as N- and C-exteins; see Figure 1A). In nature, we find so-called contiguous inteins, encoded by a single gene, as well as split inteins, encoded by two separate genes. In this case, we speak of N- and C-terminal intein fragments (Figure 1B). These fragments must first form a complex to become splice-competent; therefore, the affinity between the intein fragments clearly plays an important role in defining the splicing competency of split inteins. Regardless whether contiguous or split, inteins use a common series of chemical reactions to splice themselves out of the host. For details about this process, please refer to Di Ventura and Mootz, 2019.

Figure 1. Schematic representation of the protein splicing reaction carried out by contiguous (A) and split (B) inteins.
The Int&in web server executes a machine learning algorithm to predict whether a certain split site will give rise to two intein fragments able to assemble a functional intein competent of protein splicing. We call such sites active. Those that give rise to a split intein unable to splice are called inactive.
This explains why we chose the name "Int&in": to signify the two intein fragments (Int, in) coming together.
Why would one want to predict active split sites in inteins?
Inteins are used in protein engineering for various applications such as protein semisynthesis, labeling, and circularization (Topilina and Mills, 2014; Di Ventura and Mootz, 2019; Wang et al., 2022). In particular, split inteins enable the ligation of separate polypeptides (it could entire proteins, or protein domains or even small motif, such as nuclear localization signals, degrons etc) with no or minimal scarring -that is, without the addition or with the addition of only few amino acids originally absent from the protein of interest-, reducing the chance of compromising the function of the final protein product (Wang et al., 2022).
For instance, we recently developed a method called SiMPl that uses split intein-mediated reconstitution of enzymes that confer resistance towards antibiotics to select pure populations of cells carrying two distinct constructs with a single antibiotic in bacteria and mammalian cells (Palanisamy et al., 2019; Palanisamy et al., 2021).
Albeit there are many naturally split inteins, researchers have strived (and continue to do so) to create new split inteins either out of contiguous ones, or starting from split inteins, with the specific aim to obtain two intein fragments with low affinity for each other, which would require interacting partners to come in physical proximity and become splice-competent. This has allowed for the creation of so-called conditional inteins where the interaction between the intein fragments was triggered by an external input, such as a small chemical or light (Mootz and Muir, 2002; Böcker et al., 2019). Split inteins split at artificial new sites giving rise to two fragments having low affinity for each other were also used to develop methods to detect protein-protein interactions (Yao et al., 2020). Finally, in protein semisynthesis, where one intein fragment is chemically synthesised, it is also beneficial to have as short a fragment as possible. While one could resort to inteins naturally asymmetrically split, with one fragment being particularly short, having a method to predict where to split any intein of choice to obtain short N- or C-fragments would be very powerful.
So far, split sites in inteins have been heuristically determined...
With Int&in there is no need anymore to do trial-and-error experiments!
The algorithm
Int&in is based on a machine learning algorithm that predicts with high accuracy active and inactive split sites in inteins. It uses a logistic regression classifier on a number of sequence and structural properties to evaluate each amino acid of a given intein sequence for its potential to be an active split site. The model was trained on a dataset created by us of 126 split sites generated using the gp41-1, Npu DnaE and CL inteins and validated using 97 split sites extracted from the literature. It achieves an accuracy of 0.79 and 0.78 for the training and validation sets, respectively.
To know more about the details behind the algorithm please refer to Schmitz et al., 2023.
Before using the algorithm we advise you to read the tutorial.
Enjoy splitting your favourite intein!
Funding




Inteins are "intervening proteins": proteins found within other proteins, albeit transiently. As a matter of fact, inteins eventually excise themselves from their precursors in a process known as protein splicing (Hirata et al., 1990; Kane et al., 1990). This is an autocatalytic process, meaning that it does not need any co-factor, which concludes itself with the formation of a peptide bond between the two polypeptides initially bordering the intein (referred to as N- and C-exteins; see Figure 1A). In nature, we find so-called contiguous inteins, encoded by a single gene, as well as split inteins, encoded by two separate genes. In this case, we speak of N- and C-terminal intein fragments (Figure 1B). These fragments must first form a complex to become splice-competent; therefore, the affinity between the intein fragments clearly plays an important role in defining the splicing competency of split inteins. Regardless whether contiguous or split, inteins use a common series of chemical reactions to splice themselves out of the host. For details about this process, please refer to Di Ventura and Mootz, 2019.
Figure 1. Schematic representation of the protein splicing reaction carried out by contiguous (A) and split (B) inteins.
The Int&in web server executes a machine learning algorithm to predict whether a certain split site will give rise to two intein fragments able to assemble a functional intein competent of protein splicing. We call such sites active. Those that give rise to a split intein unable to splice are called inactive.
This explains why we chose the name "Int&in": to signify the two intein fragments (Int, in) coming together.
Why would one want to predict active split sites in inteins?
Inteins are used in protein engineering for various applications such as protein semisynthesis, labeling, and circularization (Topilina and Mills, 2014; Di Ventura and Mootz, 2019; Wang et al., 2022). In particular, split inteins enable the ligation of separate polypeptides (it could entire proteins, or protein domains or even small motif, such as nuclear localization signals, degrons etc) with no or minimal scarring -that is, without the addition or with the addition of only few amino acids originally absent from the protein of interest-, reducing the chance of compromising the function of the final protein product (Wang et al., 2022).
For instance, we recently developed a method called SiMPl that uses split intein-mediated reconstitution of enzymes that confer resistance towards antibiotics to select pure populations of cells carrying two distinct constructs with a single antibiotic in bacteria and mammalian cells (Palanisamy et al., 2019; Palanisamy et al., 2021).
Albeit there are many naturally split inteins, researchers have strived (and continue to do so) to create new split inteins either out of contiguous ones, or starting from split inteins, with the specific aim to obtain two intein fragments with low affinity for each other, which would require interacting partners to come in physical proximity and become splice-competent. This has allowed for the creation of so-called conditional inteins where the interaction between the intein fragments was triggered by an external input, such as a small chemical or light (Mootz and Muir, 2002; Böcker et al., 2019). Split inteins split at artificial new sites giving rise to two fragments having low affinity for each other were also used to develop methods to detect protein-protein interactions (Yao et al., 2020). Finally, in protein semisynthesis, where one intein fragment is chemically synthesised, it is also beneficial to have as short a fragment as possible. While one could resort to inteins naturally asymmetrically split, with one fragment being particularly short, having a method to predict where to split any intein of choice to obtain short N- or C-fragments would be very powerful.
So far, split sites in inteins have been heuristically determined...
With Int&in there is no need anymore to do trial-and-error experiments!
The algorithm
Int&in is based on a machine learning algorithm that predicts with high accuracy active and inactive split sites in inteins. It uses a logistic regression classifier on a number of sequence and structural properties to evaluate each amino acid of a given intein sequence for its potential to be an active split site. The model was trained on a dataset created by us of 126 split sites generated using the gp41-1, Npu DnaE and CL inteins and validated using 97 split sites extracted from the literature. It achieves an accuracy of 0.79 and 0.78 for the training and validation sets, respectively.
To know more about the details behind the algorithm please refer to Schmitz et al., 2023.
Before using the algorithm we advise you to read the tutorial.
Enjoy splitting your favourite intein!
Funding